
Re-Adjudicating Avacopan
The FDA names names and moves to remove avacopan from the market


Critical Commentary takes a deep dive into one argument on a systemic problem in autoimmune drug development - trial design, regulatory strategy, industry incentives, or the gap between evidence and practice.
On April 27, 2026, CDER issued a Notice of Opportunity for a Hearing (NOOH) proposing to withdraw approval of avacopan (Tavneos); the Federal Register notice followed on April 30. The FDA’s previously semi-public concerns about endpoint readjudication and hepatotoxicity have now become a formal allegation that ChemoCentryx’s NDA relied on manipulated primary-endpoint data and untrue statements of material fact, which “rendered the results [of the ADVOCATE study] biased and unreliable” and “irrevocably compromises the credibility of the study results.” ChemoCentryx (now an Amgen subsidiary) has until June 1st to request a hearing and June 29th to submit additional data and analysis. Today’s Critical Commentary will run a little long as I am hoping to present the facts as we know them in their entirety.
The FDA’s Case for Withdrawal
To save you the trouble of reading all 24 pages of the NOOH, I’ll summarize the accusations the FDA has made. These are based on internal ChemoCentryx and Medpace documents, contemporaneous emails, and Amgen’s own August 2025 written response to the agency. As of this writing, Amgen has said it “continues to believe that TAVNEOS demonstrates effectiveness and a favorable benefit–risk profile,” but to date they have not refuted the following chronology.
For brief background: the ADVOCATE trial randomized 331 patients with severe ANCA-associated vasculitis to avacopan or a prednisone taper with a background of rituximab or cyclophosphamide. The two primary endpoints were remission at week 26 (non-inferiority) and sustained remission at week 52 (superiority). The latter was non-negotiable; apparently the FDA “informed ChemoCentryx in 2016 that TAVNEOS would not be considered for approval unless the ADVOCATE study demonstrated superiority on week 52 sustained remission.” Put a pin in that; it will matter soon.
According to the FDA, on November 5, 2019, the company managing data for the ADVOCATE study (Medpace) signed off on database lock, which means the previously blinded data could now be viewed. According to the analysis plan, two ChemoCentryx employees (Director of Biostatistics Huibin Yue and Chief Medical Officer Pirow Bekker) were allowed to see the unblinded data for the purpose of conducting a quality control review “for completeness and internal consistency.”
On November 8, the topline efficacy tables were transferred to Bekker. The week 52 sustained remission analysis had failed, with a two-sided p-value of 0.1025. What happened next, in the FDA’s words:
“After being informed that there was nothing wrong with either the data or the analysis, Drs. Bekker and Yue looked for cases that could alter the study results to indicate TAVNEOS achieved a statistically superior outcome on the primary endpoint of sustained remission.”
On November 9, Bekker wrote to Yue:
“Regarding sustained remission at Week 52 it is, of course, of paramount importance that the data are correct (especially the p-value for superiority). Chao and you must verify these results ASAP. We cannot afford to miss a superiority outcome here.”
Three days later, Medpace completed a blinded data quality check that came back “with no findings” and the external statistical consultant validated the analysis. At this point, the data were confirmed correct, the analysis was confirmed correct, and the p-value was confirmed to be 0.1025. Thus far, nothing truly untoward has happened.
The targeted search. On November 12, Yue ran an unblinded query of the database for subjects with a Birmingham Vasculitis Activity Score of zero who had received glucocorticoids in the four weeks before the week 26 or week 52 assessments. This is starting to sound bad. The FDA minces no words when describing this choice:
“In the absence of any evidence of erroneous data, sponsor personnel used unblinded data specifically to find subjects whose readjudication would change the results of the trial. The evidence shows that the goal of this behavior was to alter the results of the original analysis. If the original analysis had been positive, Drs. Bekker and Yue would likely not have searched for subjects to readjudicate.”
The pre-emptive p-value. Their search ultimately identified 9 patients who had were not in remission, 6 from the avacopan group and 3 from the prednisone group. Bekker and Yue (perhaps reasonably!) believed that these patients were wrongly adjudicated. Yue next asked the consultant Chao Wang to rerun the primary endpoint analysis on a hypothetical dataset, where 5 of the 6 avacopan patients and 0 of the 3 prednisone patients somehow flipped their outcomes to sustained remission. On November 13, 2019, Dr. Wang replied that the essential 52 week primary endpoint would be superior, were those patients to flip outcomes. This is starting to raise serious concerns.
A pause for reflection. The asymmetry of the nine selected subjects is worth dwelling on, because including three prednisone-arm cases feels like a sign of good faith. The FDA did not find this convincing. They point out in the NOOH that two of the three control-arm patients had terminated the trial early and the third received glucocorticoids in a way that precluded a remission determination on its own. By construction, the agency notes:
“the three control arm patients selected for readjudication could not have made a difference in the analysis of the superiority claim at week 52.”
The FDA is saying the 3 control arm patients were window dressing. We cannot know this for certain, but they are suggesting that Bekker and Yue chose those control-arm patients because they would not flip their outcomes. The avacopan patients, on the other hand, could change.
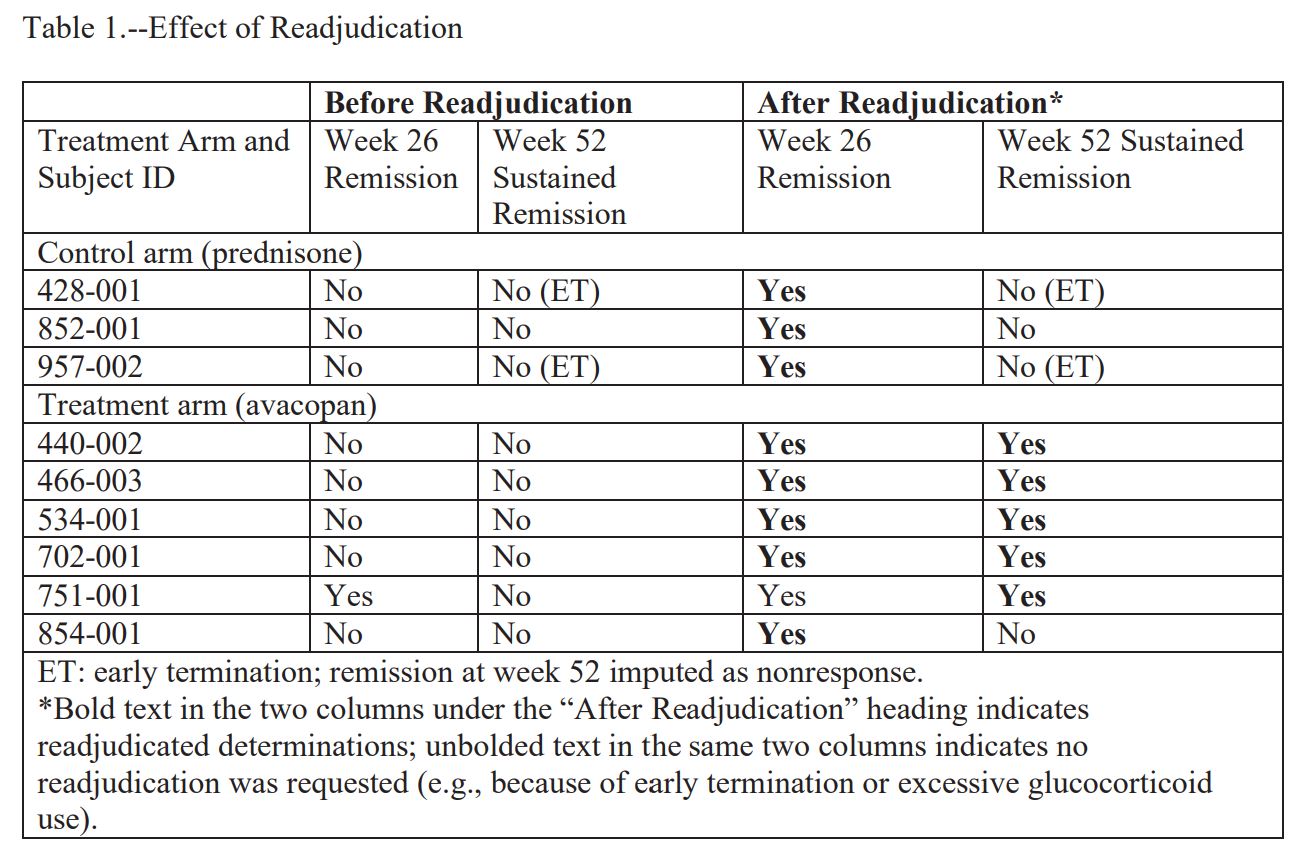
The call. On November 14, Bekker and Medpace sent the adjudication committee chair David Jayne a “patient profile” package for each of the nine subjects. According to the FDA, he reviewed each chart and reclassified five of the six avacopan subjects as in sustained remission. None of the three controls were reclassified at week 52, possibly for the aforementioned structural reasons. It is worth noting here that the FDA has not suggested that Jayne knew this was untoward; as far as I know, all parties agree he remained blinded and acted in good faith. The FDA also does not think this salvages it:
“Bias in the selection of subjects for readjudication could not be mitigated by the fact that Dr. Jayne conducted the readjudications in a blinded fashion.”
The FDA is saying that this was all pre-ordained. Whether or not Jayne knew who was in which group was immaterial, because the only way this could land was in avacopan’s favor.
The submission. On November 20, the database was locked for a second time, this time with the re-adjudicated p-value of 0.0132. That p-value, and only that p-value, was submitted to the FDA. When FDA reviewers asked about the timeline in a December 2020 information request, the response described “the freeze of the study database, which occurred on 20 November 2019” as a single event. The November 5 lock, the failed analysis, the unblinded targeted search, and the readjudications were not disclosed. The FDA letter is direct about what this concealment accomplished:
“The applicant never disclosed the readjudications that occurred after the initial database lock and that they were prompted by both an unblinded review of the study database and knowledge that the readjudications would change the study result from not statistically significant to statistically significant.”
On October 7, 2021, the FDA approved avacopan (tavneos). The agency’s account of what its own reviewers were told, and would have done if told the truth, is striking. The Division Director who recommended approval, the letter writes,
“…did not know that the facts on which he was relying were untrue, that absent manipulation, the study results were not statistically significant on the superiority analysis at week 52, and the procedures in place to ensure trial quality were violated. Had the Division Director known these facts, he would not have recommended approval.”
This is all very bad and it feels like the FDA has largely made up its mind about what to do.
Is the FDA right that it doesn’t work?
Let’s put aside the regulatory question for a moment. The broader scientific question of whether avacopan is beneficial in ANCA associated vasculitis is a little more complicated.
Bekker and Yue only flipped a few patients, and we can easily roll back the clock to see what would have happened had they not. If you strip them out and reverse the cosmetic week 26 reclassifications on the prednisone arm, which didn’t affect week 52 anyway, you get a sustained remission rate of approximately 63% on avacopan versus 55% on prednisone, an absolute difference of about 7-8%. That would still favor avacopan, though it may not meet regulatory muster.
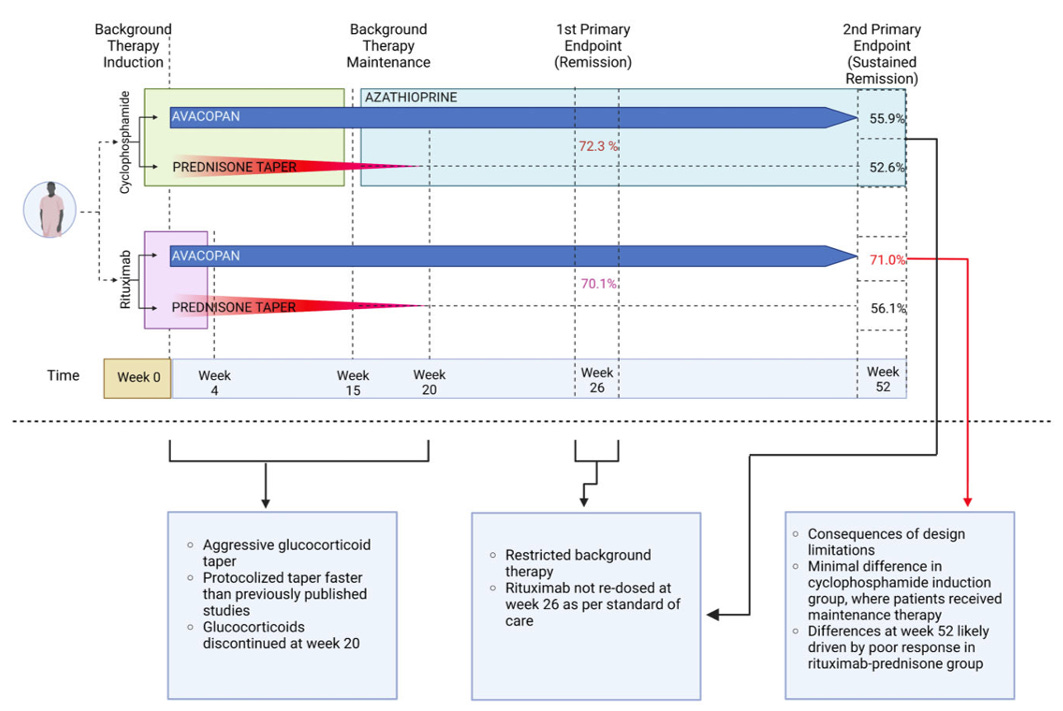
I like to share this graphic during talks, which is from a paper I published a few years back. In the paper we broke down the trial by avacopan vs. control and rituximab maintenance vs cyclophosphamide. The benefit appears to be driven by the patients who were randomized to the control group and received rituximab for induction. Those patients did not receive maintenance therapy (at all!) and clearly fared worse than those who did in the other arms. Two conclusions follow from that:
Not-treating people from week 20 to week 52 is a bad idea, and probably drove some of the perceived benefit of the drug
Giving people avacopan was obviously doing something, because those people would have flared if it did not work
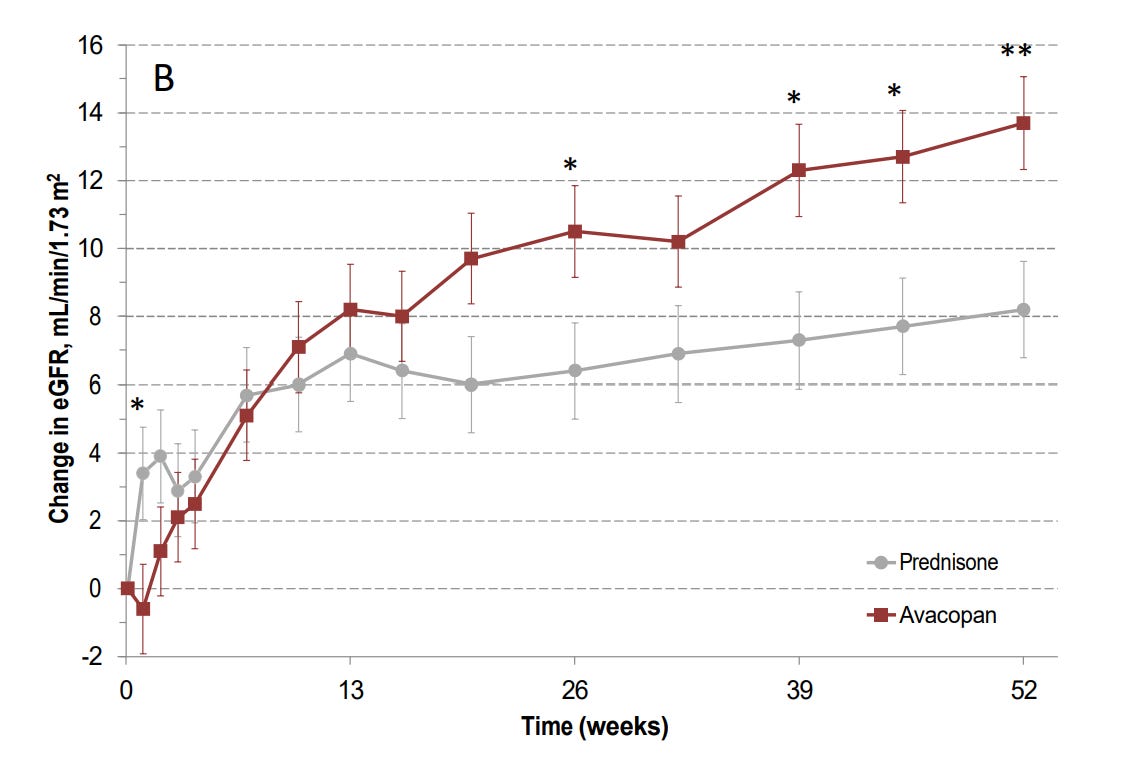
Sustained remission is also not the only metric that matters. I would argue that preventing people from going on dialysis is a substantially more important outcome measure. The ADVOCATE trial was not powered to assess progression to ESRD, but the GFR data that was not altered by Yue and Bekker also suggests a meaningful benefit from the drug. I would put an asterisk on the week 52 data because of the aforementioned problems with maintenance therapy, but I find the week 26 data pretty convincing:
Finally, we also have patient reported outcomes from the ADVOCATE trial. Patients who were randomized to received avacopan had better outcomes at both week 26 and week 52, and as far as we know those data were never touched by Bekker and Yue. An avacopan-based, glucocorticoid-sparing regimen produced better patient-reported outcomes than a protocolized prednisone taper. All of these outcomes matter a great deal, and I still believe avacopan has efficacy in ANCA associated vasculitis.
But is it safe?
So far I have been ignoring the other big story here, which is that on March 31, the FDA issued a Drug Safety Communication describing 76 postmarketing drug induced liver injury cases. Seventy-four involved a serious outcome: 54 patients were hospitalized and 8 died. Seven met criteria for biopsy-confirmed vanishing bile duct syndrome (VBDS), which is a terrifying set of words you’ve probably never seen together in a sentence before this mess emerged.
I prescribe avacopan regularly and have never seen anything like this, but therein lies the problem with rare safety signals. We typically identify them with passive reporting systems, which are complicated to interpret, to say the least. First, they suffer from the classic denominator problem. The FDA has told us all of the cases that have been reported (the numerator), but what should the denominator there be? All the prescriptions that have occurred? All of those that occurred in industrialized countries with active reporting? It’s all very nebulous.
Second, they do not come with a comparator. For all we know, this is picking up on some risk signal that is inherent to ANCA associated vasculitis. I find that difficult to believe, especially given that I have never seen VBDS and rarely see bad liver disease from ANCA associated vasculitis itself. Still, data like this cannot answer the critical question: “What is the magnitude of this risk?”
Additionally, there seems to be a remarkably strong geographic bias for these cases. Of the 76 DILI cases, most were reported from Japan (n=66), followed by the United States (n=5), Europe (n=4), and Canada (n=1). Uptake of avacopan in Japan may have been faster than in the U.S., but given the size of the populations in question it seems very unlikely that uptake and reporting culture could entirely account for it. I suspect there are some pharmacogenomic components to this that have yet to be fully characterized, which would make for a much more nuanced safety assessment (ie similar to allopurinol hypersensitivity syndrome, where some populations have HLA alleles that put them at much higher risk).
I think the jury is still out on safety. It is fair to say the safety profile in 2026 looks materially worse than the safety profile in 2021, and I have been informing all my patients of this risk. So far, nobody who hears “FDA has asked it to be withdrawn from the market” and “vanishing bile duct syndrome” has opted to start therapy with avacopan.
